ПОДРОБНАЯ ИНФОРМАЦИЯ
Заявку на получение дополнительной информации по этому проекту можно заполнить здесь.
|
Наименование инновационного проекта «Разработка новой технологии получения соединений индольного ряда» |
|
Рекомендуемая область пременения Химическая промышленность: |
|
Назначение, цели и задачи проекта Целью настоящего проекта изобретения является разработка усовершенствованного способа получения соединений индольного, которые могут найти применение в качестве полупродуктов в синтезе лекарственных препаратов. Данная цель предполагает решение следующих задач: - оптимизацию процесса за счет применения безопасных и легкодоступных исходных соединений; - увеличение выхода конечных продуктов за счет использованиея принципиально нового катализатора - комплекса циклогексаноноксимата натрия с ДМСО |
|
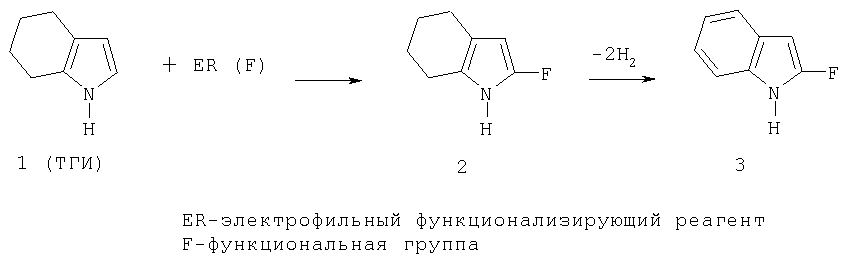
Краткое описание заменяемого процесса или решаемой проблемы Соединения индольного ряда широко представлены в живой природе, среди них - важнейшие регуляторы физиологических процессов (серотонин, мелатонин, триптофан и др.), индольные алкалоиды, в том числе нашедшие широкое применение в медицине (резерпин, стрихнин, бревиколлин, аймалин, винкамин, физостигмин и др.), синтетические лекарственные препараты (индопан, индометацин и др.), гормоны, ферменты (триптофаназа), галюциногены. Индол служит исходным продуктом в промышленном синтезе гетероауксинов (3-индолилуксусной и 3-индолилмасляной кислот), триптофана, используется в парфюмерной промышленности для создания душистых композиций в качестве фиксатора запаха. 4,5.6,7-Тетрагидроиндол (ТГИ) 1 может найти применение для синтеза индола и его труднодоступных 2-функционализированных производных. В отличие от самого индола, который атакуется электрофильными реагентами (ER) исключительно в положение 3, ТГИ, будучи по своей химической структуре пирролом, функционализируется в аналогичных реакциях селективно по положению 2 (схема 1).
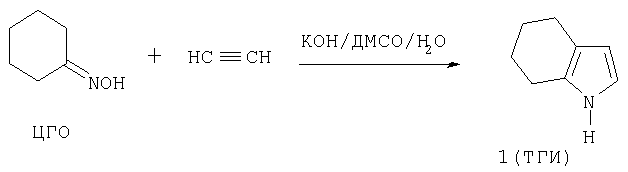
Схема 1 Дегидрированием 2-функционализированных тетрагидроиндолов 2 получают 2-функционализированные индолы 3 (И.Е.Демидов, И.Н.Буянов, И.И.Левина, Т.К.Ефимова, Т.А.Кожик, Н.Н.Суворов, Физ. хим. проблемы химических производств, М.: Московский хим. технологич. институт, 1990, с.182-183; И.Е.Демидов, И.Н.Буянов, Н.Н.Суворов, 10-ая молодежная конференция по синтет. и природ, физиол. актив. соед., Тезисы докладов, Ереван, 1990, с.42). Известен способ получения ТГИ взаимодействием циклогексаноноксима (ЦГО) с ацетиленом в присутствии гидроксида калия в водном диметилсульфоксиде (12% Н2О) под давлением ацетилена (10 атм) при 120°С (схема 2):
Схема 2 Для выделения ТГИ реакционная смесь промывается водой, экстрагируется диэтиловым эфиром, эфирные экстракты вновь многократно промываются водой и после этого осушаются карбонатом калия. Высушенные эфирные экстракты перегоняются в вакууме и закристаллизовавшийся дистиллят очищается перекристаллизацией из октана. Выход ТГИ 74%, конверсия ЦГО 50% (Авт. свид. СССР №518493, С целью повышения конверсии ЦГО в той же реакции предложен способ получения ТГИ, в котором в качестве катализатора использованы гидроокиси рубидия или тетрабутиламмония. Однако при этом оказалось необходимым повысить давление ацетилена до 16 атм (Авт. свид. СССР №620486, Описано получение ТГИ реакцией ЦГО с ацетиленом под давлением 1.2-1.5 ати в среде ДМСО - диоксан (1:1) в присутствии гидроксида калия при температуре 110-120°С и скорости подачи ацетилена 30 л/ч в течение 4-6 ч. Для выделения ТГИ используется диэтиловый эфир и перекристаллизация из гексана, гептана или октана. Выход ТГИ 45-50% (А.И.Михалева, Б.А.Трофимов, А.Н.Васильев, хим. гетероцикл. соед., 1979, №2, с.197-199). При проведении реакции при атмосферном давлении (90-95°С, 2-2.5 ч) выход ТГИ снижается до 45%. Недостатком этого способа является невысокий выход целевого продукта. Сообщалось, что реакцию могут катализировать также гидроксиды лития и натрия, однако, конверсия ЦГО и выходы целевого ТГИ при этом низки (10-20%) (А.И.Михалева, А.Н.Васильев, Б.А.Трофимов, Ж. орган. хим., 1981, т.17, в.9, с.1977-1988). Наиболее близким по технической сущности к предлагаемому изобретению является способ получения ТГИ взаимодействием ЦГО с ацетиленом в системе гидроксид калия - ДМСО при атмосферном давлении ацетилена, мольном соотношении ЦГО:КОН=1:0.3-0.4, массовом соотношении ЦГО: ДМСО=0.08 и продолжительности контакта реагентов 12-16 ч с перемешиванием реакционной смеси только в течение первых 0.5-2 ч. В этих условиях выход ТГИ и конверсию ЦГО удалось повысить (93.5 и 95%, соответственно) (Авт. свид. СССР №1705284, Особо следует остановиться на том, что в данном способе реакционная смесь перемешивается только первые 0.5-2 ч (из общих 12-16 ч), что неизбежно приводит к появлению большого градиента концентраций ацетилена по высоте реактора с максимальным его содержанием в поверхностном слое реакционной смеси и с минимальным его содержанием (или отсутствием) в нижней части реактора, что принципиально осложняет контроль за ходом реакции (поскольку в каждой точке реактора создаются разные концентрации реагентов и продукта), неизбежно снижает скорость процесса (поскольку реакция идет не в кинетической, а в диффузионной области), создает условия для нежелательного образования N-винилтетрагидроиндола (поскольку в верхних слоях реакционной смеси, где создается максимальная концентрация ТГИ, имеется и наибольшая концентрация ацетилена). Главное же - отсутствие перемешивания существенно снижает безопасность процесса, так как возникает не только градиент концентраций, но и градиент температуры, и в поверхностном слое реакционной смеси, где концентрация ацетилена наивысшая, процесс за счет большой теплоты реакции (81.0 ккал/моль) может выйти из-под контроля и вызвать взрывное разложение ацетилена (О.Н.Темкин, Т.К.Шестаков, Ю.А.Трегер. Ацетилен. Химия. Механизмы реакций. Технология. М.: Химия, 1991, с.353-361). Кроме того, фундаментальным недостатком способа-прототипа, как и способов-аналогов, использующих в качестве катализатора гидроксиды щелочных металлов, является необратимое превращение катализатора в ацетаты за счет известной реакции гидроксидов щелочных металлов с ацетиленом и водой по схеме 3 (Б.А.Трофимов. Гетероатомные производные ацетилена. Новые полифункциональные мономеры, реагенты, и полупродукты, М.: Наука, 1981, с. 14):
Схема 3 Эта побочная реакция приводит к снижению концентрации катализатора, а стало быть, и скорости основной реакции уже на начальных стадиях процесса. Она также является причиной накопления водорода в газовой фазе над реакционной смесью, что вызывает снижение парциального давления ацетилена и, как результат, дальнейшему падению скорости основной реакции. Кроме того, в процессе используют для выделения ТГИ большие количества (800 мл на моль исходного ЦГО) экстрагента (дихлорметана или эфира, первый - ядовит, второй - пожаро-взрывоопасен). Экстракты промывают водой от ДМСО, который при этом безвозвратно теряется (регенерация разбавленных водных растворов ДМСО энергоемка). Отмытые экстракты сушат прокаленным поташом, который неизбежно адсорбирует экстрагенты, исходный и конечный продукты, а следовательно, не подлежит регенерации. Кроме своей очевидной нетехнологичности, эта стадия приводит еще к безвозвратным потерям экстрагентов и продукта. |
|
Краткое описание предлагаемого технологического процесса Цель проекта достигается использованием принципиально нового катализатора - комплекса циклогексаноноксимата натрия с ДМСО 4, состава 1:1
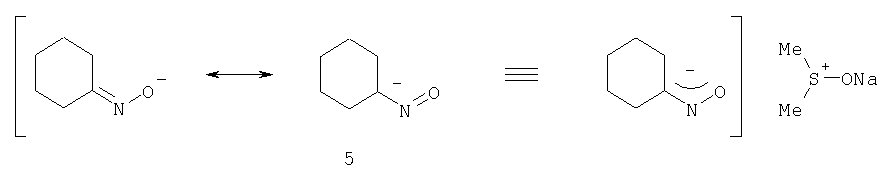
С учетом известной способности ДМСО комплексно связывать катионы щелочных металлов (Дж. Гордон. Органическая химия растворов и электролитов. М.: Мир, 1979, с.311-319), электронное строение комплекса может быть представлено формулой 5.
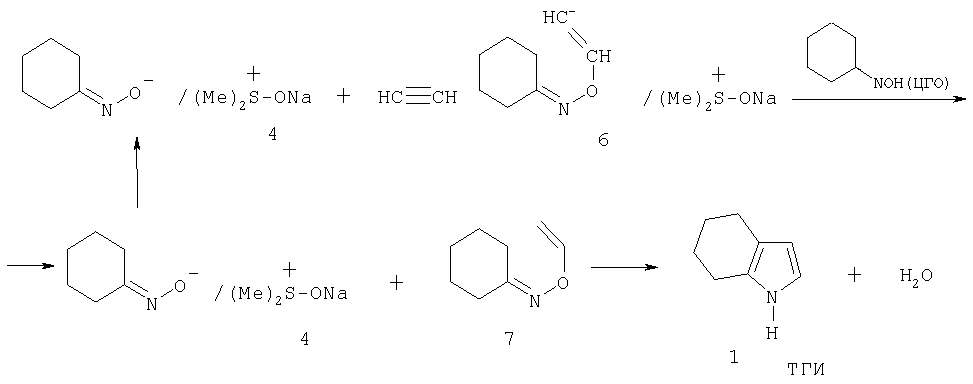
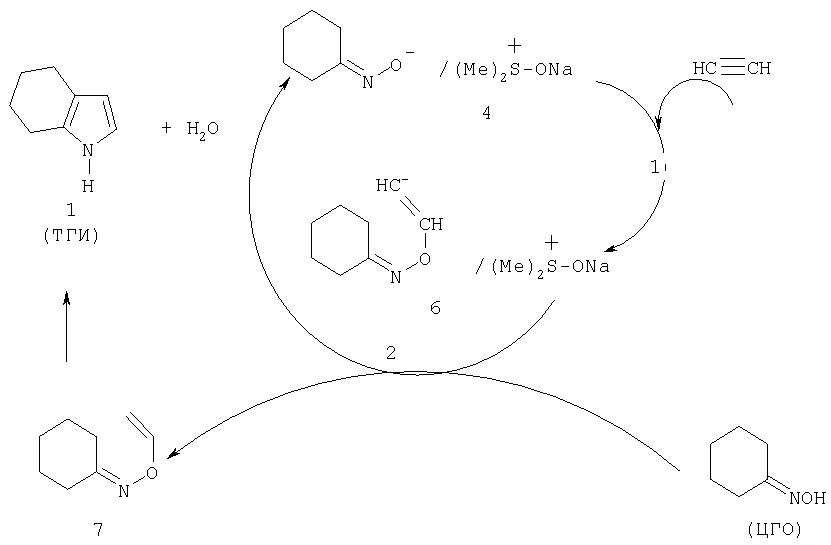
Из приведенной структуры 5 видно, что контактная ионная пара циклогексаноноксимата натрия разделяется молекулой ДМСО, который связывает катион натрия в объемный сульфоксониевый катион с распределенным (менее концентрированным) положительным зарядом. В результате электростатическое катион-анионное взаимодействие в новой ионной паре ослабляется, и анион циклогексаноноксима становится более нуклеофильным, т.е. более реакционноспособным по отношению к молекуле ацетилена. В силу существующего обмена со средой реагирующий циклогексаноноксим-анион в ходе реакции с ацетиленом уступает место новой молекуле циклогексаноноксима с образованием интермедиата - О-винилоксима 7 (Б.А.Трофимов, А.И.Михалева. N-винилпирролы. Новосибирск: Наука, 1984, с.24-28), который далее перегруппировывается в целевой продукт - ТГИ, и каталитический цикл возобновляется (схема 4):
Схема 4 Более наглядно каталитический цикл может быть изображен схемой 5:
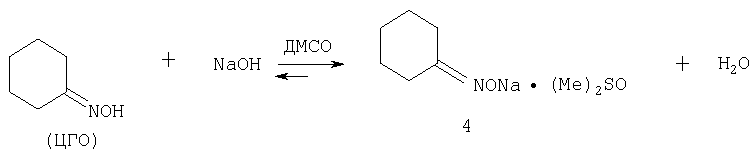
Схема 5 Ключевой стадией процесса является внедрение молекулы ацетилена в каталитический комплекс 4 с образованием промежуточного карбаниона 6 (реакция 1), который, принимая протон от новой молекулы ЦГО, дает интермедиат 7 (О-винилоксим), далее перегруппировывающийся в ТГИ. Катализатор 4 может быть получен взаимодействием ЦГО с гидроксидом натрия в ДМСО при температуре 80-130°С (схема 6).
Схема 6 Для более полного сдвига равновесия в сторону образования комплекса 4 выделяющаяся вода удаляется в виде азеотропа с подходящим азеотропным агентом (бензолом, циклогексаном, толуолом, гептаном, октаном, этанолом и др., предпочтительно толуолом). Для удобства дозирования катализатора он может быть приготовлен в виде раствора в смеси ЦГО-ДМСО, который остается гомогенным при температуре 80-130°С (в зависимости от концентрации в нем комплекса 4). Преимуществом предлагаемого способа является то, что для его осуществления катализатор по сравнению с катализатором прототипа (КОН) готовится из безводного и более дешевого гидроксида натрия, тогда как коммерческий гидроксид калия, применяемый в способе-прототипе, всегда содержит около 15% воды, что замедляет процесс и ускоряет побочные реакции. Другим преимуществом предлагаемого способа является меньший расход каталитического прекурсора (NaOH): в предлагаемом способе 3-10% (NaOH) от массы ЦГО против 17-25% (КОН) в способе-прототипе. Главным преимуществом предлагаемого способа является то, что при указанной меньшей концентрации катализатора достигается 4-х кратное сокращение времени процесса: 1.5-3 часа вместо 12-16 ч в способе-прототипе. Еще одним существенным преимуществом нового способа является то, что он позволяет работать с более концентрированными растворами, т.е. использовать меньшие количества активного растворителя (ДМСО): при реализации процесса по новому способу концентрация ЦГО в реакционной смеси составляет 13-35% против 8% в способе-прототипе, следовательно, используются в 3 раза более концентрированные реакционные смеси, что существенно повышает интенсивность процесса и снижает время и энергозатраты, необходимые на регенерацию ДМСО. К дополнительным существенным преимуществам предлагаемого способа относится то, что он обеспечивает возможность прямой дистилляции ДМСО из реакционной смеси в вакууме после нейтрализации ее углекислым газом. При этом отгоняется основная часть (80-90%) ДМСО, а компоненты оставшегося сырого продукта (ТГИ, ЦГО, ДМСО) разделяются экстракцией небольшими порциями водного слабо концентрированного (10-20%) раствора NaOH и толуола. Сырой продукт может быть предварительно отделен от нейтрализованного катализатора (карбоната натрия) перегонкой в вакууме, либо перегонкой при атмосферном давлении с водой или водяным паром. Перегонка с водой может также проводиться в невысоком вакууме. Таким образом, решается принципиальная для данной технологии проблема регенерации дорогостоящего растворителя ДМСО (нерешенная в способе-прототипе), из процесса устраняются токсичные (дихлорметан) и пожаровзрывоопасные (диэтиловый эфир) экстрагенты. В общем виде предлагаемый способ получения ТГИ осуществляется следующим образом: в нагретый (100-130°С) ДМСО, продутый азотом для удаления кислорода и углекислого газа, прибавляется в токе азота нагретый (100-130°С) раствор катализатора - комплекса 4, приготовленного из NaOH и циклогексаноноксима в ДМСО (с отгонкой воды в виде азеотропа с толуолом или бензолом, циклогексаном, гептаном, октаном, этанолом). Предпочтительные концентрации компонентов в приготовленной таким образом реакционной смеси (катализаторном растворе): циклогексаноноксим - 10-30%, более предпочтительно 13-17%; NaOH - 0.1-3.0%, более предпочтительно 0.5-1.5%. В реакционную смесь при температуре 100-140°С, предпочтительно 120-130°С, пропускается ацетилен со скоростью, обеспечивающей его конверсию 90-100%, предпочтительно 95%. Через 1.5-4 часа после достижения конверсии ЦГО, равной 60-95%, предпочтительно 70-80%, подача ацетилена прекращается, реакционная смесь выдерживается при температуре реакции 0.5-1 ч до полного израсходования растворенного в ней ацетилена, продувается азотом, нейтрализуется (СО 2 или SO2, NH4 Cl, HCl) и подвергается перегонке в вакууме. Сначала отгоняется 80-90% ДМСО, который после азеотропной осушки снова может быть использован в процессе, затем сырой продукт, содержащий ДМСО (30-50%), ТГИ (30-40%) и ЦГО (10-20%). Эта фракция интенсивно перемешивается с равным объемом толуола и 0.2-0.4 объема водного раствора NaOH (10-20%). После расслоения водная щелочная фаза отделяется, добавляется новая порция водного NaOH (0.1-0.2 объема от объема толуольного слоя) и экстракция повторяется. Процесс экстракции заканчивается, когда в толуольном слое определяются лишь следовые количества ЦГО. Из толуольного слоя отгоняется толуол, при этом происходит одновременная азеотропная осушка остающегося в кубе ТГИ. Перегонкой кубового остатка в вакууме получают чистый ТГИ (т.к. ТГИ имеет т.пл. 50°С, температура в конденсате и приемнике поддерживается в пределах 60-70°С), выход 95-97% при конверсии ЦГО 70-90%. Из водно-щелочных экстрактов отгоняется вода, а остаток, содержащий ДМСО, NaOH и циклогексанонгоксимат натрия в виде комплекса с ДМСО, используется далее для приготовления катализаторного раствора. Следующие примеры иллюстрируют предлагаемую технологию. Пример 1. А. Приготовление катализаторного раствора. Смесь Катализатор был получен в чистом виде, для этого смесь Найдено, %: С 45.45, Н 7.68, N 6.30, Na 10.97, S 14.76. C 8H16NNaO2S. Вычислено, %: С 45.05, Н 7.56, N 6.57, Na 10.78, S 15.03. Титрование комплекса 4: навеска М=0.0431?1000/(0.105?1.9)=217 Теоретически для формулы 4 - 213. В спектре ЯМР 1Н комплекса 4 (Bruker-400DPX, 400.13 МГц, ДМСО-d 6, Значительные сильнопольные всех сигналов 13С циклогексаноноксимного фрагмента дополнительно подтверждают усиление анионного характера оксимной части комплекса 4 за счет образования оксимата натрия и его комплексообразования с ДМСО. В ИК спектре (KBr, см-1) комплекса 4 имеется узкая интенсивная полоса при Б. Синтез 4,5,6,7-тетрагидроиндола (ТГИ). Полученный катализаторный раствор помещают в стеклянный реактор емкостью 20 мл с магнитной мешалкой, разбавляют 7.00 мл ДМСО, продувают азотом, затем ацетиленом, реактор соединяют с ацетиленовой бюреткой (ацетиленовая трубка находится над поверхностью катализаторного раствора), и реакционную смесь нагревают (130°С) при перемешивании, фиксируя во времени скорость поглощения и общее количество прореагировавшего ацетилена (кинетическая кривая поглощения ацетилена приведена на фиг.1). Через каждые полчаса реактор продувают свежим ацетиленом. По истечении 2 ч после поглощения 250 мл ацетилена (84% от объема, необходимого для полного превращения взятого количества ЦГО в ТГИ), подачу ацетилена прекращают, реакционную смесь переносят в перегонную колбу, пропускают в нее 100 мл CO 2 и смесь перегоняют в вакууме. Отгоняют 8.41 мл ДМСО (50-67°С / Из щелочных экстрактов (6.1 мл) отгоняют воду и небольшие количества толуола в вакууме (?100 мм Hg, температура нагревателя 90 -98°С) и получают Пример 2. В условиях примера 1, в том же реакторе, используя те же загрузки (суммарно 10 мл ДМСО, Пример 3. В условиях примера 1, в том же реакторе, с теми же загрузками реагентов и катализатора, но при температуре 100°С, проводя реакцию в течение 3 ч, выделяют, как описано в примере 1, Пример 4. В условиях примера 1, в том же реакторе, с теми же загрузками реагентов, но с катализатором, соответствующим Пример 5. В условиях примера 1, в том же реакторе, используя тот же объем ДМСО (10 мл), при той же концентрации катализатора (соответствующего Пример 6. В стеклянный реактор с магнитной мешалкой объемом 200 мл загружают в токе азота 70 мл ДМСО и 45 мл катализаторного раствора, приготовленного из Пример 7. Как описано в примере 6, в том же реакторе, но используя Пример 8. В стеклянный реактор с механической мешалкой объемом Пример 9. В стальной реактор с механической мешалкой емкостью Пример 10. Синтез проводят в реакторе, описанном в примере 6, с теми же загрузками ДМСО и ЦГО, но с катализатором, приготовленным из Пример 11. В реактор, описанный в примере 6, загружают 70 мл ДМСО и 80 мл катализаторного раствора, приготовленного из |
|
Технико-экономическое обоснование применения инновационной технологии Себестоимость тетрагидроиндола, получаемого по предлагаемой технологии (с использованием нового катализатора) ниже, чем при его получении по традиционным технологиям в 10 раз. По каталогам зарубежных фирм (FLUKA, ALDRIGE) стоимость 1 грамма ТГИ составляет 100 евро. По новой технологии стоимость составляет около 10 евро. Применение новой технологии позволяет снизить эксплуатационные расходы, исключить поставку дорогостоящих реагентов (за счет сокращение времени реакции с 13-15 до 1.5-3 часов), что экономит от 15 млн. рублей в год. |
|
Технико-экономические показатели трудо-энерго-природосбережения нового процесса Преимуществом предлагаемой технологии является то, что для его осуществления катализатор по сравнению с катализатором прототипа (КОН) готовится из безводного и более дешевого гидроксида натрия, тогда как коммерческий гидроксид калия, применяемый в способе-прототипе, всегда содержит около 15% воды, что замедляет процесс и ускоряет побочные реакции. Другим преимуществом предлагаемой технологии является меньший расход каталитического прекурсора (NaOH): в предлагаемом способе 3-10% (NaOH) от массы ЦГО против 17-25% (КОН) в способе-прототипе. Главным преимуществом предлагаемого технологии является то, что при указанной меньшей концентрации катализатора достигается 4-х кратное сокращение времени процесса: 1.5-3 часа вместо 12-16 ч в способе-прототипе. Еще одним существенным преимуществом нового способа является то, что он позволяет работать с более концентрированными растворами, т.е. использовать меньшие количества активного растворителя (ДМСО): при реализации процесса по новому способу концентрация ЦГО в реакционной смеси составляет 13-35% против 8% в способе-прототипе, следовательно, используются в 3 раза более концентрированные реакционные смеси, что существенно повышает интенсивность процесса и снижает время и энергозатраты, необходимые на регенерацию ДМСО. К дополнительным существенным преимуществам предлагаемого способа относится то, что он обеспечивает возможность прямой дистилляции ДМСО из реакционной смеси в вакууме после нейтрализации ее углекислым газом. При этом отгоняется основная часть (80-90%) ДМСО, а компоненты оставшегося сырого продукта (ТГИ, ЦГО, ДМСО) разделяются экстракцией небольшими порциями водного слабо концентрированного (10-20%) раствора NaOH и толуола. Сырой продукт может быть предварительно отделен от нейтрализованного катализатора (карбоната натрия) перегонкой в вакууме, либо перегонкой при атмосферном давлении с водой или водяным паром. Перегонка с водой может также проводиться в невысоком вакууме. Таким образом, решается принципиальная для данной технологии проблема регенерации дорогостоящего растворителя ДМСО (нерешенная в способе-прототипе), из процесса устраняются токсичные (дихлорметан) и пожаровзрывоопасные (диэтиловый эфир) экстрагенты. |
|
Новые потребительские свойства продукции - Высокий выход тетрагидроиндола (до 90%) |
|
Качественные характеристики, предъявляемые к сырью и материалам Продукция соответствует государственным стандартам |
|
Стадия и уровень разработки Технология прошлая ряд испытаний на пилотных производствах Восточной Сибири. |
|
Предлагаемые инвестиции |
|
Рынки сбыта |
|
Возможность и эффективность импортозамещения |
|
Возможность выхода на мировой рынок |
|
Срок окупаемости (в месяцах) |
|
Дата поступления материала 18.09.2007 |
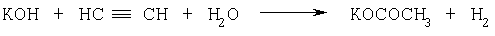
Инновации и люди

У павильонов Уральской выставки «ИННОВАЦИИ 2010» (г. Екатеринбург, 2010 г.)

Мероприятия на выставке "Инновации и инвестиции - 2008" (Югра, 2008 г.)

Открытие выставки "Малый бизнес. Инновации. Инвестиции" (г. Магнитогорск, 2007 г.)

Демонстрация разработок на выставке "Малый бизнес. Инновации. Инвестиции" (г. Магнитогорск, 2007 г.)
2007–2025 © Инновации - Бизнесу. Инновации и инвестиции в прорывные технологии